To, że tu jesteście oznacza najprawdopodobniej, że szukacie diagnozy dla swojego Dziecka. Albo już ją macie – CLN2 – jesteście przerażeni, zrozpaczeni, załamani…
Chcemy choć trochę oswoić Was z tym, co ta choroba za sobą niesie. Ale przede wszystkim chcemy dodać Wam nadziei i siły. Będziemy mówić o niej „po ludzku”, nie tylko naukowo i bardzo specjalistycznie. Głowa do góry – jest zadane do wykonania!
Ważne!!! CLN2 można leczyć
Od 2017 roku dostępne jest leczenie hamujące postęp choroby (w Europie, w Polsce jeszcze nie). Trwają też zaawansowane prace nad kolejnymi terapiami [zobacz].
Piszemy o objawach i przebiegu choroby, żeby:
- po pierwsze, pomóc szybciej ją diagnozować,
- po drugie, by pokazać, jak istotne jest szybkie włączenie leczenia.
Demencja dziecięca – brzmi jak oksymoron, ale tak, jest taka choroba. Inne jej nazwy to cerolidolipofuscynoza neuronalna (w skrócie: CLN, NCL) lub zespół Battena.
W zależności od typu, a jest ich kilkanaście, choroba ta objawia w różnym wieku i ma zróżnicowany przebieg. Generalnie, im później pojawią się pierwsze objawy, tym wolniej postępuje, niemniej w każdym przypadku sukcesywnie odbiera wcześniej zdobyte umiejętności, sprawność a ostatecznie podstawowe funkcje życiowe.
| Pierwsze objawy w okresie… | Typ CLN |
| noworodek/niemowlę | 10 |
| małe dziecko (>6 miesięcy) | 1, 2, 5, 6, 7, 8, 14 |
| dziecko szkolne | 1, 2, 3, 5, 6, 7, 8, 10, 12 |
| młody dorosły | 1, 2, 4, 6, 10, 11, 13 |
| Deficyt enzymów lizosomalnych | 1, 2, 10, 13 |
| Deficyt białek nieenzymatycznych | 3, 4, 5, 6, 7, 8, 11, 12, 14 |
Typy CLN charakterystyczne dla danej grupy wiekowej
Źródło: Schulz A, Kohlschütter A. NCL Disorders: Frequent Causes
of Childhood Dementia. Iran J Child Neurol. 2013 Winter; 7(1):1-8.
Wszystkie CLN są ultrarzadkie, metaboliczne i mają podłoże genetyczne. Wśród nich najczęściej występuje właśnie CLN2. Zazwyczaj manifestuje się ok. 3 roku życia i w naturalnym przebiegu jest okrutna – zabiera dzieci w wieku 8-12 lat.
Czemu ultrarzadka?
O chorobie ultrarzadkiej mówimy, kiedy 1 chory przypada na grupę 50 tys. mieszkańców. Więcej na ten temat piszemy w zakładce Koń czy zebra. Polskich dzieci chorych na CLN2 jest kilkadziesiąt, co roku pojawiają się 2-5 nowych przypadków. Nie znamy dokładnych statystyk, bo aktualnie brakuje rejestru takich danych (pracujemy nad tym w ramach Rejestr chorych). Większość chorych zaraz po diagnozie wyjeżdża z kraju, by podjąć leczenie tam, gdzie jest ono refundowane (krajem docelowym są głównie Niemcy).
Czemu metaboliczna?
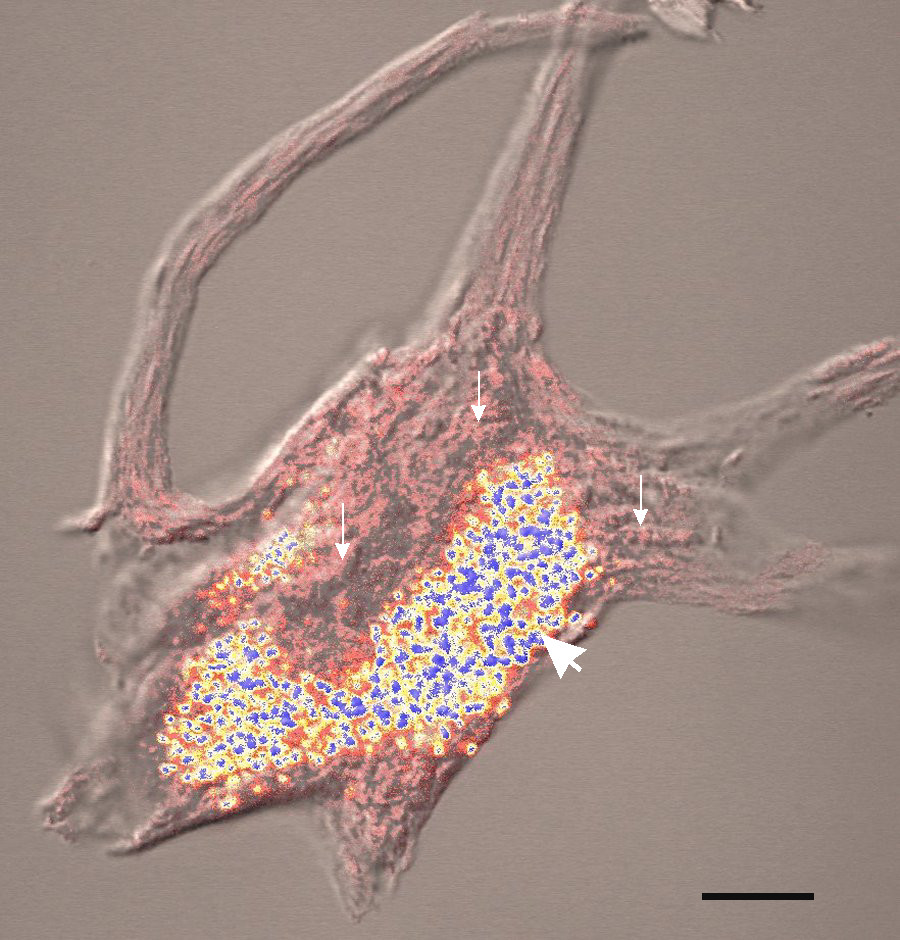
Żródło: https://www.med.unc.edu
CLN cechuje nieprawidłowy przebieg procesów metabolicznych w orga nizmie. Jest on spowodowany brakiem lub niewystarczającym poziomem enzymu TPP1 (tripeptydylopeptydaza 1), który odpowiada za oczyszczanie komórek z odpadów „poprodukcyjnych”. W każdej komórce znajdują się lizosomy, a w lizosomach powinien działać enzym. Jeśli jest go za mało, materiał spichrzeniowy (złogi) gromadzi się i z czasem uszkadza komórkę.
Uszkodzenia te najszybciej manifestują się w mózgu, zaraz potem w oczach. Postępujące zmiany w układzie nerwowym powodują kolejne, coraz poważniejsze zaburzenia procesów życiowych, o czym szerzej w zakładce Diagnoza i przebieg.

Źródło: https://www.cln2family.com
Czemu genetyczna?
Zaburzenie metaboliczne w postaci niewystarczającej produkcji enzymu TPP1 to efekt błędu genetycznego – mutacji w genie TPP1. CLN2 to zatem choroba jednogenowa a mutacja dotyczy genu kontrolującego syntezę białek enzymatycznych. Jej dziedziczenie następuje w sposób autosomalny recesywny. Co to oznacza?
Z genetycznego punktu widzenia możemy być właścicielem:
- prawidłowego genu, wówczas jesteśmy zdrowi;
- wadliwego genu, wówczas chorujemy;
- nosicielem mutacji genu, wówczas nie chorujemy, ale „noszoną” mutację możemy, choć nie musimy, przekazać swojemu dziecku.
Problem pojawia się wtedy, kiedy nosicielstwo zostanie przekazane dziecku przez oboje rodziców. Spotkanie dwójki nosicieli to trochę jak „wygrana” na loterii – zdarza się bardzo rzadko, wręcz ultrarzadko. I nawet, jeśli jesteśmy nosicielami, to statystycznie rzecz biorąc, przy każdej ciąży mamy aż 75% szans na to, by nasze dziecko urodziło się zdrowe, w tym 50% na to, że będzie to bezobjawowy nosiciel (Grafika 3).
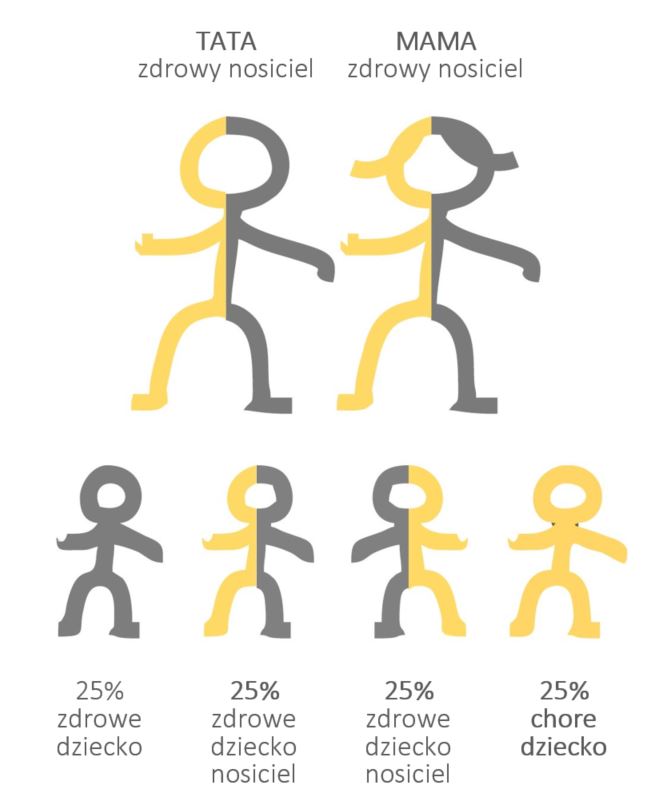
Źródło: Opracowanie własne.
Niestety statystyka bywa wierutnym kłamstwem. Spotykamy rodziny, które zmagają się z chorobą nawet trójki dzieci. Konfiguracje mogą być przeróżne: 1 chore i 1 zdrowe dziecko (nawet z ciąży bliźniaczej) czy 2 chorych dzieci i 1 zdrowe. Może być i tak, że wszystkie dzieci są zdrowe i nie wiedzą, że są nosicielami błędu genetycznego i w przyszłości mogą przekazać go swoim dzieciom.
To, czy jesteś nosicielem można sprawdzić!
Tu dotykamy bardzo ważnego tematu – badań na nosicielstwo. Jeśli w rodzinie pojawi się CLN2 (czy też inna choroba genetyczna), badania takie powinni przejść:
- rodzeństwo chorego dziecka, przy czym badanie dokonywane jest w ramach usług poradni genetycznej dopiero po uzyskaniu przez nie pełnoletności;
- rodzeństwo rodziców chorego dziecka, a gdy okażą się nosicielami, badaniom powinni poddać się ich partnerzy i dzieci;
- dziadkowie chorego dziecka z obu stron, bowiem badanie wskaże, która część rodziny jest zagrożona nosicielstwem i powinna podać się badaniom.
Mutacja „krąży” w rodzinie i może być przekazywana kolejnym pokoleniom. Nigdy nie wiadomo, czy jej nosiciel nie spotka drugiej zmutowanej „połówki”, co będzie grozić chorobą kolejnego dziecka.
Ten fakt często bywa bagatelizowany, dlatego podkreślamy, że badanie na nosicielstwo może uchronić przed wielkim dramatem, jakim niestety jest choroba genetyczna.
Mity:
* rodzice dzieci z CLN2 na pewno są spokrewnieni;
* w rodzinie z 4 dzieci tylko 1 z nich będzie chore;
* błąd genetyczny powstał po stronie matki;
* w mojej rodzinie wszyscy byli zdrowi, ta choroba to wina drugiej strony.
Mechanizm dziedziczenia jest niezależny od płci – CLN2 dotyka zarówno osoby płci męskiej, jak i żeńskiej. Wykonanie badań w kierunku CLN2 możliwe jest na wczesnym etapie ciąży. O możliwościach diagnostycznych po urodzeniu dziecka szerzej w zakładce Diagnoza i przebieg.











